SAMSON Web is live: create, predict, render, and share molecular systems directly in your browser!
Today, we are incredibly proud to introduce SAMSON Web: a browser-based molecular design environment.
No installation.
No plugin.
No account required.
Open SAMSON Web, create or import a molecular system, work on it directly in the browser, and share it.
Not a screenshot.
Not a video.
A real interactive 3D molecular document.
With the full context.
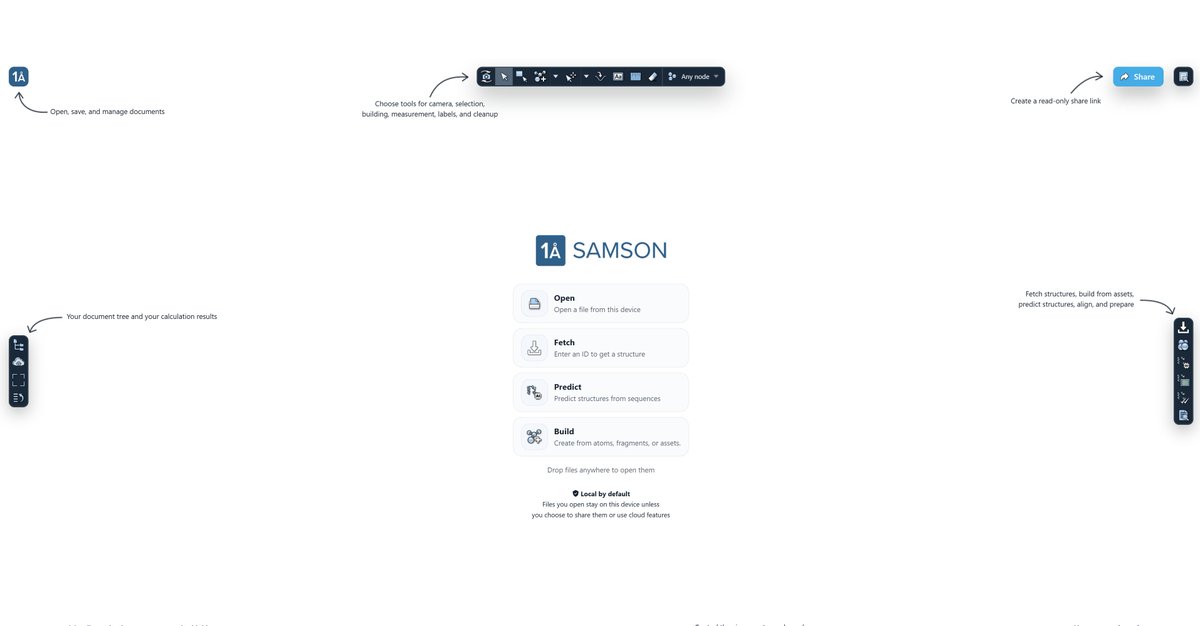
With SAMSON Web, you can start in many ways:
• open molecular files from your device
• import and export common formats such as PDB, MOL2, and more
• fetch structures from databases
• predict structures (currently with AlphaFold 2, Boltz-2, and Chai-1x)
• build from atoms, fragments, and assets
• import cloud job results
And once your system is in the browser, you can work on it: select, measure, prepare, align, minimize, annotate, edit, create a new version, and share again.
Because one of the most important parts of SAMSON Web is sharing interactive documents.
Scientific communication still relies too much on molecular images or movies.
But molecular systems are three-dimensional. And they have context, annotations, associated data, versions, and intent.
With SAMSON Web, you can share an interactive molecular document that preserves context in seconds.
A collaborator, student, reviewer, customer, or colleague can open it in the browser, rotate it, zoom in, inspect annotations, associated logs and data, understand the model directly, and even edit their own copy. Remote collaborators, even during a Zoom or Teams session, can now explore designs as they want, edit, and send you back their changes.
SAMSON Web also helps you create high-quality visuals directly in the browser:
• advanced rendering effects
• custom backgrounds
• 3D skyboxes
• path tracing
• advanced materials
• snapshots and render-ready scenes
Modern molecular design produces much more than coordinates: prediction results, alignments, tables, metadata, trajectories, reports, annotations, and intermediate files.
SAMSON documents preserve it all, and SAMSON Web also includes a Data Explorer to explore and edit rich scientific data next to molecular structures.
The goal is to make molecular documents richer and more useful: scientific workspaces where structure, data, calculations, and interpretation live together.
You start locally by default (files you open stay on your device unless you choose to share them or use cloud features).
You do not need an account to create and share.
Creating an account unlocks versioned documents and history, permission controls, and access to your data from anywhere.
SAMSON Web is live. You can start in seconds.
The link is in the first comment.
#OneAngstrom #SAMSON #MolecularDesign #ComputationalBiology #DrugDiscovery #ProteinDesign #StructuralBiology #MaterialsScience #Nanoscience #AlphaFold #Boltz #Chai #MolecularVisualization #ScientificComputing #AIForScience