Targeted enzyme discovery using metal-coordination mining
1 Metal-coordination mining uses atomic-level active-site geometry (not overall sequence similarity) to search predicted structures for specific metalloenzyme functions, enabling targeted discovery within huge, diverse superfamilies.
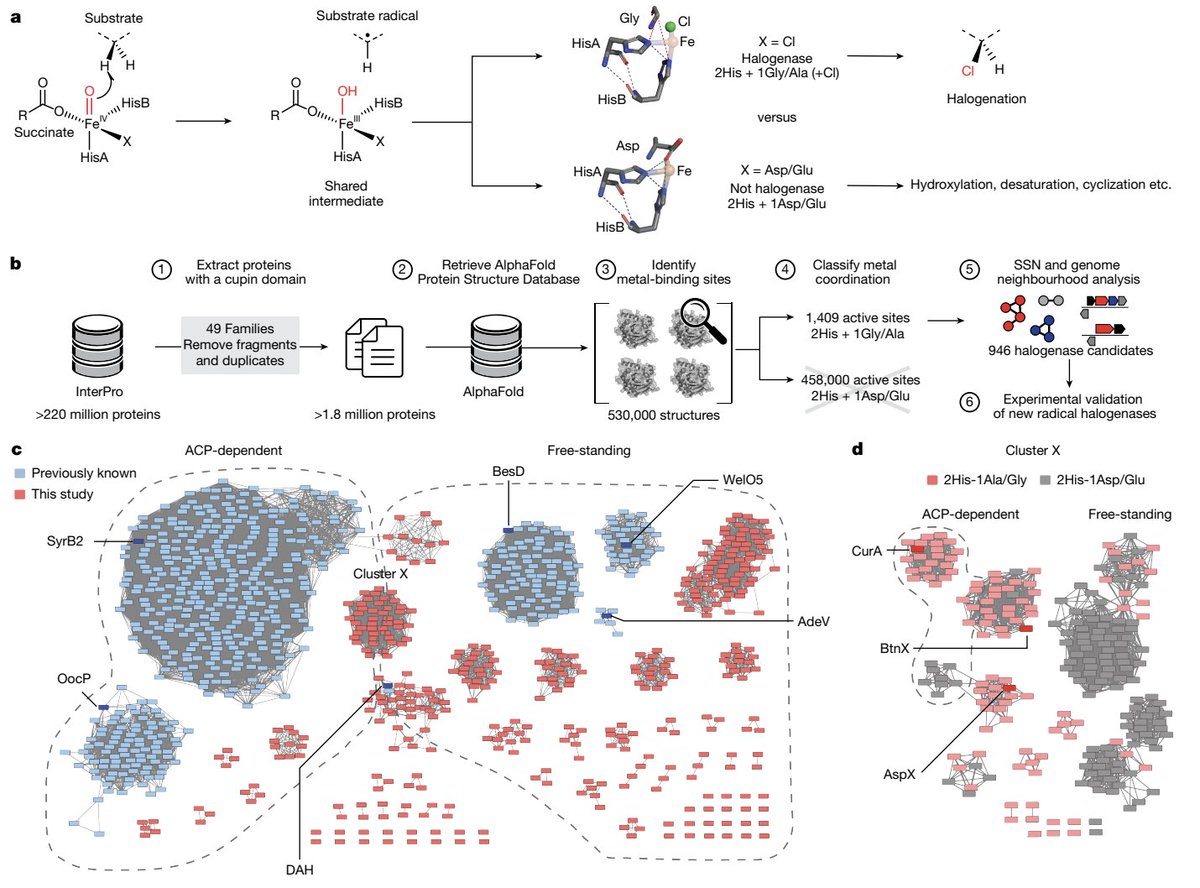
2 The key mechanistic insight: FeII/αKG-dependent radical halogenases require an open metal coordination site for halide binding, so the canonical 2His-1Asp/Glu facial triad is replaced by a 2His-1Gly/Ala motif; this absence of Asp/Glu becomes a minimal structural signature for halogenation.
3 The pipeline scales efficiently by searching 3D motifs (effectively N^1) rather than pairwise sequence comparisons (N^2), making it practical for database-scale mining where subtle residue differences are otherwise hard to detect.
4 Applied to InterPro AlphaFold2 DB: from ~220M sequences, the authors extracted ~1.8M cupin-domain proteins, retrieved ~530,814 AF2 structures, identified ~458,000 predicted 2His metal sites, and then pinpointed 946 candidates with 2His-1Gly/Ala (putative radical halogenases).
5 A sequence-similarity network built from these candidates recapitulated all known FeII/αKG halogenase families and expanded the landscape dramatically: 70 previously unrecognized clusters spanning broad phylogenetic space, including multiple new eukaryotic-associated groups (e.g., a much larger DAH-related cluster than BLAST reveals).
6 Experimental validation focused on a newly identified “cluster X” with mixed genomic contexts (ACP-associated and apparently free-standing). Genome neighborhood analysis guided substrate hypotheses rather than relying on sequence alone.
7 AspX (from Vibrio campbellii) was shown to be a free amino-acid halogenase that selectively converts L-aspartate to 3S-chloro-L-aspartate (kcat ~33.3 min−1, Km ~0.64 mM), extending known free-substrate halogenation to a negatively charged amino acid; it can also install Br and N3 with alternative anions.
8 BtnX (from Dinoroseobacter shibae “killer plasmid”) was linked by gene context to biotin uptake and validated as a biotin halogenase producing 2R-chlorobiotin with very tight binding (Km < 2 μM), consistent with low marine biotin availability; product stereochemistry was supported by crystallography.
9 BtnX is unusually substrate-promiscuous for this enzyme class: it halogenates many non-native carboxylate-containing molecules (from fatty acids to dyes to peptides) as long as a propionate-like head group is present, enabling access to diverse α-halo acids relevant to synthesis and late-stage functionalization.
10 Structural basis of promiscuity: crystal structures show specific H-bonding that anchors the substrate carboxylate near the reactive center, while the remainder of the substrate extends into a solvent-exposed channel with mostly nonspecific interactions; a single active-site mutation (G117D/E) switches BtnX from halogenation to hydroxylation, highlighting how metal-coordination rules can also guide enzyme reprogramming.
💻Code: doi.org/10.5281/zenodo.19737…
📜Paper: doi.org/10.1038/s41586-026-1…
#ComputationalBiology #Bioinformatics #EnzymeDiscovery #Metalloenzymes #AlphaFold #StructuralBioinformatics #Biocatalysis #NaturalProducts #ProteinEngineering
1
25
2,222