quoting this because it’s a lens into something most never think about
what IS a “good” model. Rohit and i want two very diff things here.
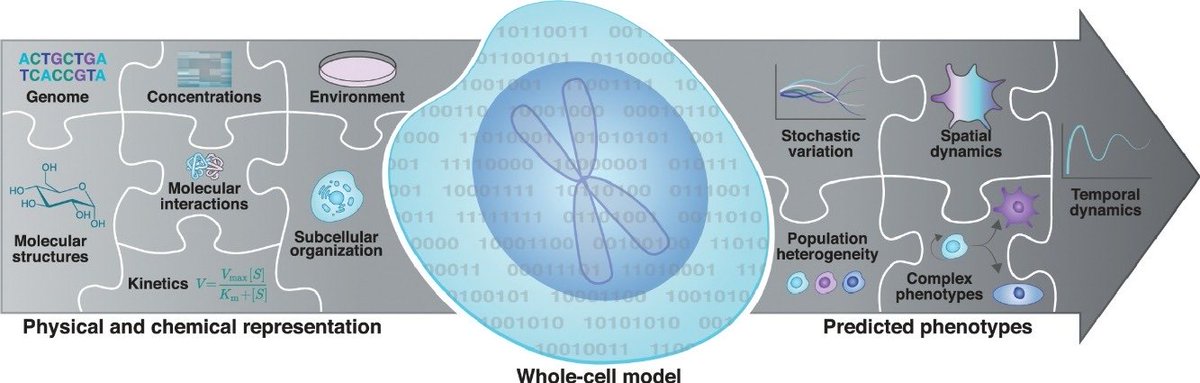
OP wants a *provably correct* virtual cell. right from biophysics.
to me, that’s…useless.
let’s learn why, because it MATTERS.
🧵
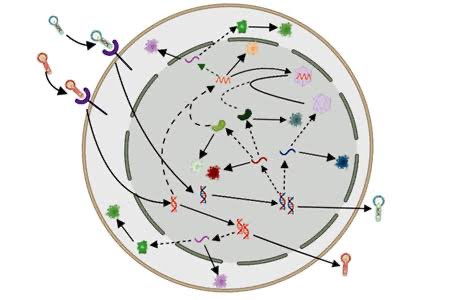
The point I am trying to make is that learning an energy landscape from empirical cellular imaging trajectories, a pre-requisite for training an accurate model for cell state dynamics, is bound to break the assumption of Markovianity. In other words, your energy function(al) will have tiny errors due to experimental resolution and/or artifacts of your apparatus which will inhibit adequate replication of the true dynamics.
This is the same issue that has plagued the MD community for several decades now. You could argue that Alphafold is a counter example but you need to scratch below the surface only a little bit to realize why protein structure prediction does not help with inferring protein motion.
Here are a few practical model-agnostic Markovianity tests you could try to see the light:
1. Rodríguez‐Girondo, M., & de Uña‐Álvarez, J. (2012). A nonparametric test for Markovianity in the illness‐death model. Statistics in Medicine, 31(30), 4416-4427.
2. Berezhkovskii, A. M., & Makarov, D. E. (2018). Single-molecule test for Markovianity of the dynamics along a reaction coordinate. The journal of physical chemistry letters, 9(9), 2190-2195.
3. Willareth, L., Sokolov, I. M., Roichman, Y., & Lindner, B. (2017). Generalized fluctuation-dissipation theorem as a test of the Markovianity of a system. Europhysics Letters, 118(2), 20001.